全部
▼
搜索
熱搜:
位置:中冶有色 >
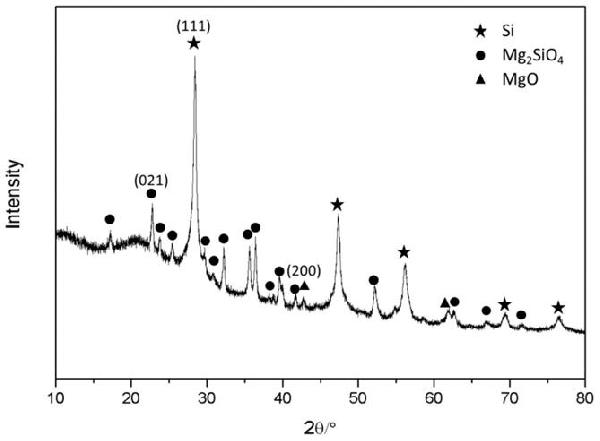
> 氧化亞硅負(fù)極材料及其制備方法與流程
 1050
編輯:中冶有色技術(shù)網(wǎng)
來源:萬華化學(xué)集團(tuán)股份有限公司
1050
編輯:中冶有色技術(shù)網(wǎng)
來源:萬華化學(xué)集團(tuán)股份有限公司
 分享 0
分享 0
 舉報 0
舉報 0
 收藏 0
收藏 0
 反對 0
反對 0
 點贊 0
點贊 0



 2025年03月20日 ~ 22日
2025年03月20日 ~ 22日 材料焊接與連接技術(shù)交流會") 2025年03月28日 ~ 30日
2025年03月28日 ~ 30日 術(shù)會議") 2025年03月28日 ~ 30日
2025年03月28日 ~ 30日 創(chuàng)新大會") 2025年03月28日 ~ 30日
2025年03月28日 ~ 30日 資源科技創(chuàng)新發(fā)展論壇") 2025年04月27日 ~ 29日
2025年04月27日 ~ 29日 












































































