全部
▼
搜索
熱搜:
 205
編輯:中冶有色技術網(wǎng)
來源:陜西恒昌鉬業(yè)有限公司
205
編輯:中冶有色技術網(wǎng)
來源:陜西恒昌鉬業(yè)有限公司
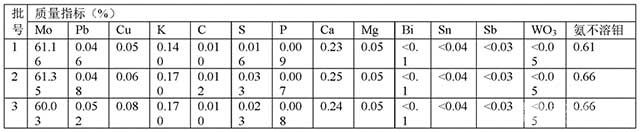
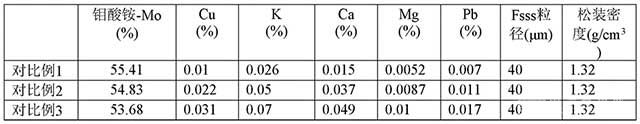
[0049]表1 焙燒鉬精礦分析化驗指標
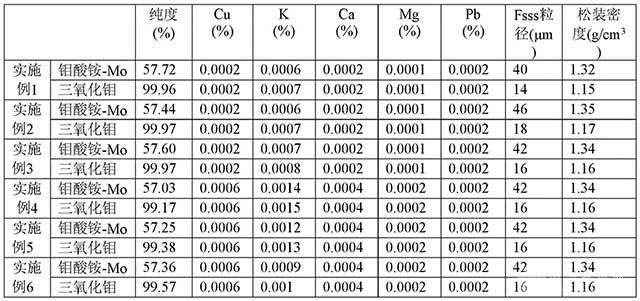
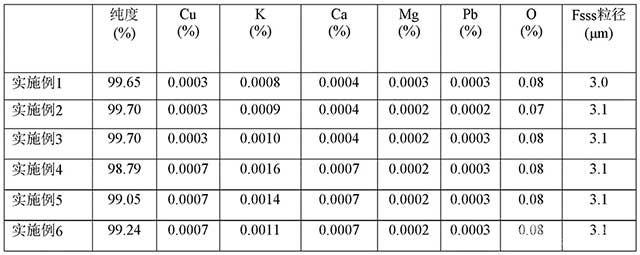
[0092]表2 實施例1~6鉬酸銨-Mo的含量和三氧化鉬純度以及雜質含量檢測
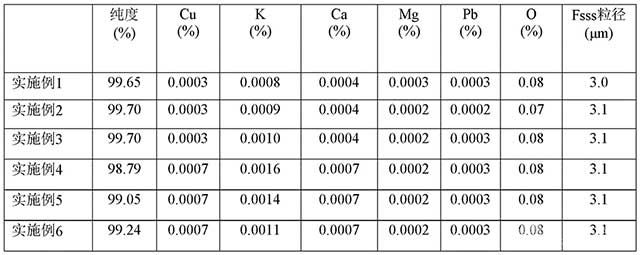
[0093]表3 實施例1~6中鉬粉的純度以及雜質含量檢測
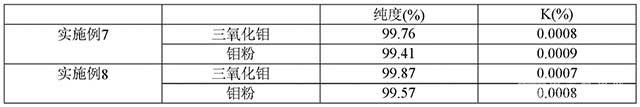

 分享 0
分享 0
 舉報 0
舉報 0
 收藏 0
收藏 0
 反對 0
反對 0
 點贊 0
點贊 0












































































 2025年03月20日 ~ 22日
2025年03月20日 ~ 22日 







