權(quán)利要求
1.基于氮摻雜的電極材料��,其特征在于��,包括以下制備步驟:
將氮稠環(huán)前驅(qū)體摻雜到氧化石墨中����,得到氮摻雜石墨烯;
將氮摻雜石墨烯負載到碳布上�,即得到氮摻雜的電極材料;
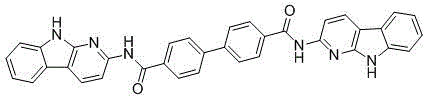
所述的氮稠環(huán)前驅(qū)體的結(jié)構(gòu)式為:
2.根據(jù)權(quán)利要求1所述的基于氮摻雜的電極材料���,其特征在于���,所述的電極材料中氮摻雜石墨烯的負載質(zhì)量為2.5-5mg/cm2。
3.根據(jù)權(quán)利要求1所述的基于氮摻雜的電極材料����,其特征在于,所述的氧化石墨的制備方法為:在冰浴條件下�,將石墨�、硝酸鈉和冰鎮(zhèn)后的濃硫酸依次加入燒瓶中�����,混合均勻后�,分批次加入高錳酸鉀�����,經(jīng)氧化反應后加入純化水和雙氧水終止反應�����,經(jīng)酸洗����、水洗、冷凍干燥后即得到氧化石墨�����。
4.根據(jù)權(quán)利要求3所述的基于氮摻雜的電極材料��,其特征在于�,分批次加入高錳酸鉀具體為分2次加入:第一次是將高錳酸鉀加入到冰浴中酸混合液中,混合均勻后,移除冰浴�,在室溫下,第二次加入高錳酸鉀�����,室溫攪拌后再移入水浴中����,在35-45℃的水浴中繼續(xù)攪拌2-3h。
5.根據(jù)權(quán)利要求4所述的基于氮摻雜的電極材料����,其特征在于,第一次加入的高錳酸鉀與第二次加入的高錳酸鉀的質(zhì)量比為1:20-25�。
6.根據(jù)權(quán)利要求1所述的基于氮摻雜的電極材料,其特征在于��,氮稠環(huán)前驅(qū)體的制備方法為:稱量2.0-2.3g 4�����,4'-聯(lián)苯二甲醛����、3.6-4.0g 9H-吡啶并[2�,3-b]吲哚-2-胺��、1.5-2g無水硫酸鈉�、2.0-2.5 g叔丁醇鈉加入到圓底燒瓶中,接著加入200-300ml無水乙腈�����,將上述燒瓶加入到水浴溫度為60-70℃的水浴鍋中���,攪拌反應6-10h,監(jiān)測反應結(jié)束后����,旋干溶劑得到的混合物采用柱層析分離,即氮稠環(huán)前驅(qū)體��。
7.根據(jù)權(quán)利要求6所述的基于氮摻雜的電極材料����,其特征在于,所述的監(jiān)測采用薄層色譜版進行展開分析監(jiān)測�����,展開劑為正己烷或石油醚:乙酸乙酯:二氯乙烷=2.5-5:2:1。
8.根據(jù)權(quán)利要求6所述的基于氮摻雜的電極材料����,其特征在于,所述的柱層析分離采用的層析液為正己烷或石油醚:乙酸乙酯:二氯乙烷=3.5-5.5:2:1��。
9.權(quán)利要求1制備的基于氮摻雜的電極材料的應用����,其特征在于,將制得的所述氮摻雜的電極材料用于雙電層超級電容器�����。
說明書
技術領域
[0001]本發(fā)明屬于超級電容器領域�,涉及電子元件的制備領域,具體涉及一種基于氮摻雜的電極材料及其在超級電容器上的應用��。
背景技術
[0002]隨著科技的進步和經(jīng)濟的快速發(fā)展�,人們對化石燃料的需求和消耗大幅度提高,成本也隨之增加�,且環(huán)境污染、全球變暖和能源資源日漸短缺等現(xiàn)象日益嚴重�����,這促進了人們對可再生新能源和高效能量轉(zhuǎn)換及存儲技術的關注;超級電容器又叫電化學電容器�����,它的能量密度高于傳統(tǒng)電容器�����,且功率密度比二次電池高出一個數(shù)量級�����,同時還具有能大電流快速充放電����,無記憶效應�����,循環(huán)壽命長�����,安全性高���,工作溫度范圍寬�,環(huán)境友好,免維護等優(yōu)點���,是現(xiàn)在炙手可熱的儲能器件��。
[0003]高性能超級電容器具有高比電容����,高功率密度和能量密度及良好的循環(huán)穩(wěn)定性�,但是純石墨烯基超級電容器仍無法滿足高性能的要求。經(jīng)研究發(fā)現(xiàn)����,石墨烯材料的比電容與理論值的差距仍然很大,純石墨烯基超級電容器的比電容大多低于200 F/g����,這主要是由于石墨烯層片間有較強的范德華力,化學還原或熱還原制得的石墨烯易發(fā)生堆疊導致石墨烯片團聚和堆疊�����,石墨烯在電解液中的分散性和浸潤性變差��,有效比表面積和離子電導率的降低導致比電容減小。因此�����,開發(fā)單層石墨烯�,對石墨烯進行化學改性,增強石墨烯與其它活性材料的協(xié)同作用是目前石墨烯電極材料研究的重要方向��。
發(fā)明內(nèi)容
[0004]本發(fā)明的目的在于提供一種基于氮摻雜的電極材料及其在超級電容器上的應用����。
[0005]由于石墨烯片層之間存在較強的π-π共軛,易發(fā)生堆疊和團聚�,自然不利于其在電解液中的分散和浸潤,在實際應用中���,為了改善石墨烯的電化學性能,本發(fā)明采用對其功能化改性��。本發(fā)明的目的可以通過以下技術方案實現(xiàn):
基于氮摻雜的電極材料�,包括以下制備步驟:
將氮稠環(huán)前驅(qū)體摻雜到氧化石墨中,得到氮摻雜石墨烯�����;
所述的氮稠環(huán)前驅(qū)體的結(jié)構(gòu)式為:
電極材料的制備:稱取10mg氮摻雜石墨烯加入到10ml體積分數(shù)為50%的乙醇水溶液中,超聲分散30-40min��,移取10ul分散液到直徑為5mm的碳布上��,在50-55℃下干燥10-12h�,即得到電極材料,其中電極材料中氮摻雜石墨烯的負載質(zhì)量為2.5-5mg/cm2�。
[0006]進一步,所述的氧化石墨的制備方法為:將三口燒瓶放入到冰浴中���,稱量4g石墨和2g硝酸鈉加入其中�����,接著倒入100ml冰鎮(zhèn)(溫度<5℃)后濃度為98%的濃硫酸�,緩慢攪拌30-40min后��,待混合均勻����,接著加入少量高錳酸鉀,攪拌混合30-40min后�,移除冰浴,在室溫下,再次加入高錳酸鉀���,攪拌60-80min�,接著放入到溫度為35-45℃的水浴中攪拌2-3h���,攪拌結(jié)束后�,向燒瓶中加入200ml純化水���,10-15min加入完畢�,最后加入20ml濃度為30%的雙氧水����,攪拌5-10分鐘,得到亮黃色的溶液�����,經(jīng)離心��、5%鹽酸進行酸洗����、純化水洗滌,最后經(jīng)冷凍干燥即得到氧化石墨�����。
[0007]分批次加入高錳酸鉀得到的氧化石墨具有更加均勻均一面內(nèi)孔以及較大的比表面積����,研究發(fā)現(xiàn),一次性加入高錳酸鉀得到的氧化石墨�,由于高錳酸鉀吸附在石墨上的含量不均勻,得到的氧化石墨其形成的面內(nèi)孔太不均一��,進而在后期摻雜氮稠環(huán)前驅(qū)體會使其摻雜不均勻���,降低其電性能���。先加入少量的高錳酸鉀進行預氧化,形成一定的面內(nèi)孔的石墨����,并附上一定量的含氧基團,再次加入高錳酸鉀后����,可在原來的面內(nèi)孔上進一步氧化碳原子進而獲得更大的面內(nèi)孔���,即增加了比表面積,且在更大的面內(nèi)孔上可獲得更多功能含氧基團���,有利于氮稠環(huán)前驅(qū)體的摻雜���,這樣做成電極材料后,電解液離子可直接進入高比表面石墨烯的內(nèi)部����,有利于電荷積累、離子擴散���,進而可以得到更高的比電容��。這樣具有較大的比表面積和孔容的電極材料做成雙電層超級電容器����,可以很好的提高雙電層超級電容器的電性能�����。
[0008]進一步���,第一次加入的高錳酸鉀與第二次加入的高錳酸鉀的質(zhì)量比為1:20-25��。
[0009]進一步����,氮稠環(huán)前驅(qū)體的制備方法為:稱量2.0-2.3g 4����,4'-聯(lián)苯二甲醛、3.6-4.0g9H-吡啶并[2�����,3-b]吲哚-2-胺���、1.5-2g無水硫酸鈉��、2.0-2.5 g叔丁醇鈉加入到圓底燒瓶中�,接著加入200-300ml無水乙腈��,將上述燒瓶加入到水浴溫度為60-70℃的水浴鍋中�,攪拌反應6-10h,監(jiān)測反應結(jié)束后�,旋干溶劑得到的混合物采用柱層析分離��,即氮稠環(huán)前驅(qū)體�。
[0010]本發(fā)明制備的氮稠環(huán)前驅(qū)體��,其結(jié)構(gòu)中的吡啶上的N可與氧化石墨上的氫形成氫鍵��,酰胺官能團和N-H也可以與氧化石墨形成氫鍵��,甚至可能形成含有氫鍵的大環(huán)���,這樣通過氫鍵和吸附作用的摻雜��,使得氮稠環(huán)前驅(qū)體在氧化石墨中更加穩(wěn)定����,進而做成的電極材料其性能更加穩(wěn)定��;
氮稠環(huán)前驅(qū)體被引入到石墨烯的炭網(wǎng)平面中����,改變了石墨烯的電子結(jié)構(gòu)和固有屬性,由于氮原子的電子排布與碳原子相似���,并且電負性較大��,氮稠環(huán)前驅(qū)體中的氮原子在電極材料中會變成電子供體��,提高炭材料表面的電荷遷移率����,摻入氮稠環(huán)前驅(qū)體后�,還會提高炭材料的潤濕性,有利于電解液離子的遷移��,電解液離子能夠更高地滲透到石墨烯材料中���,增大其可利用的雙電層面積�����,進而可以增大超級電容器的比電容����。
[0011]進一步����,所述的監(jiān)測采用薄層色譜版進行展開分析監(jiān)測,展開劑為正己烷或石油醚:乙酸乙酯:二氯乙烷=2.5-5:2:1�����。
[0012]進一步,所述的柱層析分離采用的層析液為正己烷或石油醚:乙酸乙酯:二氯乙烷=3.5-5.5:2:1�。
[0013]進一步,上述制備的基于氮摻雜的電極材料的應用�,是將制得的所述氮摻雜的電極材料用于雙電層超級電容器。
[0014]本發(fā)明的有益效果:
本發(fā)明制備的基于氮摻雜的電極材料具有良好的電導率和較高的比表面積��,以氮摻雜石墨烯電極材料作為電極��,以KOH水溶液為電解液��,組裝成雙電層超級電容器�����,在1A/g電流密度時的比電容為274F/g��;經(jīng)5000次循環(huán)后�,電容保持率為91.7%,具有良好的電性能和電學穩(wěn)定性��。
[0015]當然��,實施本發(fā)明的任一產(chǎn)品并不一定需要同時達到以上所述的所有優(yōu)點。
附圖說明
[0016]為了更清楚地說明本發(fā)明實施例的技術方案���,下面將對實施例描述所需要使用的附圖作簡單地介紹���,顯而易見地,下面描述中的附圖僅僅是本發(fā)明的一些實施例�,對于本領域普通技術人員來講,在不付出創(chuàng)造性勞動的前提下�,還可以根據(jù)這些附圖獲得其他的附圖���。
[0017]圖1中A為本發(fā)明氧化石墨的電鏡掃描圖�;B為氮摻雜石墨烯的電鏡掃描圖����;
圖2為氮稠環(huán)前驅(qū)體的制備流程圖;
圖3為氮稠環(huán)前驅(qū)體的核磁圖譜���。
具體實施方式
[0018]下面將結(jié)合本發(fā)明實施例中的附圖�,對本發(fā)明實施例中的技術方案進行清楚����、完整地描述,顯然��,所描述的實施例僅僅是本發(fā)明一部分實施例,而不是全部的實施例�����?���;诒景l(fā)明中的實施例,本領域普通技術人員在沒有作出創(chuàng)造性勞動前提下所獲得的所有其它實施例����,都屬于本發(fā)明保護的范圍。
[0019]實施例1 氧化石墨烯的制備
采用水熱法制備氧化石墨:
將三口燒瓶放入到冰浴中��,稱量4g石墨和2g硝酸鈉加入其中��,接著倒入100ml冰鎮(zhèn)(溫度<5℃)后濃度為98%的濃硫酸�����,緩慢攪拌30min后�,待混合均勻,接著加入0.5g高錳酸鉀���,攪拌混合30min后����,移除冰浴,在室溫下���,再次加入10g高錳酸鉀���,攪拌60min,接著放入到溫度為40℃的水浴中攪拌2h��,攪拌結(jié)束后����,向燒瓶中加入200ml純化水�,15min加入完畢,最后加入20ml濃度為30%的雙氧水�����,攪拌7分鐘��,得到亮黃色的溶液��,經(jīng)離心、5%鹽酸進行酸洗��、純化水洗滌��,最后經(jīng)冷凍干燥即得到氧化石墨��,如圖1中A所示�����。
[0020]實施例2 氮稠環(huán)前驅(qū)體的制備
稱量2.2g4����,4'-聯(lián)苯二甲醛、3.8g9H-吡啶并[2����,3-b]吲哚-2-胺、2g無水硫酸鈉��、2.0g叔丁醇鈉加入到圓底燒瓶中����,接著加入250ml無水乙腈,將上述燒瓶加入到水浴溫度為65℃的水浴鍋中�����,攪拌反應8h,采用薄層色譜版進行展開分析監(jiān)測�����,展開劑為正己烷:乙酸乙酯:二氯乙烷=3:2:1����,反應結(jié)束后,將燒瓶放入旋轉(zhuǎn)蒸發(fā)儀����,旋干溶劑得到的混合物采用柱層析分離,層析液為正己烷:乙酸乙酯:二氯乙烷=4:2:1����,得到了氮稠環(huán)前驅(qū)體�。反應流程如圖2結(jié)構(gòu)所示;
氮稠環(huán)前驅(qū)體結(jié)構(gòu)檢測:1 H NMR (500 MHz�, CDCl3) δ: 10.12 (s, 2H)���, 8.95(s�����, 2H)��,7.86 (d���, J=4.8 Hz�����, 2H)�,7.83 (d���, J=8.0 Hz�, 4H) ���,7.57 (d��, J=8.0 Hz���,4H) ,7.32 (d����, J=4.8 Hz�����, 2H) �����,7.07-7.21 (m���, 4H) ,6.94 (td���,J=8.0�����, 4.5 Hz��,2H)�����,6.33 (d����, J=4.8 Hz���, 2H) 如圖3所示�;HRMS calcd for C36H24N6O2 [M+H]+572.1237�����, found 572.1226�����。
[0021]實施例3 氮摻雜石墨烯的制備
在燒杯中加入500ml體積分數(shù)為50%的乙醇水溶液��,接著依次加入0.5g氧化石墨���、0.25g氮稠環(huán)前驅(qū)體����,接著將燒杯轉(zhuǎn)移至超聲儀中進行超聲分散2h(200W/250kHz)����,接著向燒杯中加入200ml氨水�����,繼續(xù)超聲分散15min���,得到的混合溶液轉(zhuǎn)移至高溫反應釜中,在185℃下反應11h����,反應結(jié)束后,經(jīng)抽濾�、去離子水洗滌至中性、最后經(jīng)冷凍干燥(冷凍干燥條件為溫度為-50℃�,真空壓力為8.1Pa,干燥時間為18-24h)���,即得到氮摻雜石墨烯�,如圖1中B所示���。
[0022]實施例4 電極材料的制備
稱取10mg氮摻雜石墨烯加入到10ml體積分數(shù)為50%的乙醇水溶液中��,超聲分散30min��,移取10ul分散液到直徑為5mm的碳布上��,在55℃下干燥11h��,即得到電極材料��,其中電極材料中氮摻雜石墨烯的負載質(zhì)量為3mg/cm2����。經(jīng)檢測�,采用RTS-8四探針測試儀測得電極材料的的電導率為143S/cm,采用Micromeritics Instrument Corporation 公司生產(chǎn)的TriStar II 3020 型比表面分析儀檢測氮摻雜石墨烯的比表面積為1412m2/g��。以氮摻雜石墨烯電極材料作為電極�����,以濃度為8mol/L KOH水溶液為電解液�,組裝成雙電層超級電容器,在1A/g電流密度時的比電容為274F/g��;在1A/g電流密度下����,經(jīng)5000次循環(huán)后,電容保持率為91.7%。
[0023]以上內(nèi)容僅僅是對本發(fā)明的構(gòu)思所作的舉例和說明�,所屬本技術領域的技術人員對所描述的具體實施例做各種各樣的修改或補充或采用類似的方式替代,只要不偏離發(fā)明的構(gòu)思或者超越本權(quán)利要求書所定義的范圍��,均應屬于本發(fā)明的保護范圍��。
全文PDF
基于氮摻雜的電極材料及其在超級電容器上的應用.pdf
聲明:
“基于氮摻雜的電極材料及其在超級電容器上的應用” 該技術專利(論文)所有權(quán)利歸屬于技術(論文)所有人����。僅供學習研究,如用于商業(yè)用途�,請聯(lián)系該技術所有人。
我是此專利(論文)的發(fā)明人(作者)
 877
編輯:中冶有色網(wǎng)
來源:深圳市今朝時代股份有限公司
877
編輯:中冶有色網(wǎng)
來源:深圳市今朝時代股份有限公司
 分享 0
分享 0
 舉報 0
舉報 0
 收藏 0
收藏 0
 反對 0
反對 0
 點贊 0
點贊 0










































































材料焊接與連接技術交流會")

 2025年03月28日 ~ 30日
2025年03月28日 ~ 30日 爐窯及耐火材料產(chǎn)學研合作高峰論壇")







